DOI: 10.31038/CST.2018323
Abstract
Tumor cell migration and invasion are critical steps in the metastatic cascade and depend on the interaction between tumor cells, the extracellular matrix (ECM) and the endothelial cells. Integrins are key receptors that link cells and ECM, acting as mechanical sensors of the cell microenvironment. Particularly, Arg-Gly-Asp (RGD)-binding integrins such as αvβ3 and α5β1 integrins are of special interest in cancer progression. Integrins also interact with growth factor receptors resulting in an important cross talking between intracellular signaling pathways. Studies have provided evidence of distinct roles for αvβ3 and α5β1 integrins during migration. Therefore, αvβ3 and α5β1 integrins became an attractive target for pharmacological inhibition in cancer therapy and metastasis prevention. Cilengitide, the first integrin inhibitor based on the RGD motif, is currently under clinical trial in cancer patients with limited success. Monoclonal antibodies to integrins also presented modest results. Therefore, efforts to achieve a better understanding on the integrin roles in cancer progression are needed. In the last few years, new mechanisms that may help to explain the lack of success of integrin inhibitors were described and are commented here.
Keywords
integrin, cancer, disintegrins
Introduction
Cancer is one of the major concerns related to human health, and metastasis, when occurs, is the main cause of deaths in patients with cancer. Despite all efforts of academic, governmental and private institutions, there are very few options to prevent or treat metastasis. [1] One of the reasons for this lack of success relays on the current limited knowledge on the cellular programs driving the process of metastasis. Recently, new mechanisms that allow prolonged tumor cell survival after loss of attachment to the ECM have been reported, including autophagy and entosis [2]. In addition to shedding some light in the knowledge of tumor progression, these mechanisms provided new targets and options for metastasis treatment. Here we review some key aspects of cell attachment/detachment to ECM by integrins in the context of tumor microenvironment. We will also comment on the results obtained so far with integrin-targeted anticancer therapy.
Tumor Microenvironment and the Integrins
In the past ten years, the microenvironment where tumor cells develop has achieved special importance mostly due to its pivotal role in tumor progression. The tumor microenvironment (TME) provides signals and several kinds of support from distinct cell types and from the extracellular matrix (ECM) [3, 4]. Signals from the TME comprise soluble factors released by stromal cells such as fibroblasts, stem and immune cells, blood vessels, products from proteolysis of ECM components and cytokines [5]. On the other hand, tumor cells release proteases, microvesicles and growth factors that affect and modify the surrounding cells and the ECM. The TME also has a key role in the resistance to therapy due to a continuous communication with tumor cells and modulation of their responses [6, 7]. Integrins are among the crucial cell surface receptors in supporting the cross talk between cells and ECM, and therefore are critically involved in tumor progression [8].
Integrins are transmembrane receptors that support the adhesion of cells to the ECM [8]. Loss of integrin adhesion usually induces cell death unless cells can find new adhesion sites. Integrin are formed by heterodimers containing one α and one β subunits with the ability to recognize ECM components such as collagen (Col), fibronectin (FN) and laminin (LM) with high affinity [9]. There are 18 α subunits and 8 β subunits that can be combined in several ways to form distinct receptors with different specificity to ECM components. For instance, α5β1 integrin is the main receptor for FN whereas vitronectin (VN) binds preferentially to αvβ3 integrin. Both α5β1 and αvβ3 integrins recognize the tripeptide RGD motif within ECM proteins; however other integrins also binds to the RGD motif such as αvβ1, αvβ5, αvβ6, αvβ8, α8β1, and the platelet fibrinogen receptor, αIIbβ3 integrin [10]. One of the most interesting feature of integrins is the fact that, despite binding the same ligand, each one has its own cell-dependent pattern of expression and plays distinct roles in cell adhesion and migration [11].
Integrins in cell adhesion and migration
Integrin clustering and activation upon ligand binding triggers intracellular signaling pathways including the activation of several kinases like the focal adhesion kinase (FAK), mitogen activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK) resulting in changes in cell behavior [8, 12]. Cell adhesion and migration are among the main cell abilities that are under strict integrin control and are crucial steps during tumor progression. Moving cells may change their integrin profile in response to modifications on the ECM milieu. In a FN-rich microenvironment, cells will depend mostly on integrins such as α5β1 and αvβ3 for motility and, despite their ability in binding to the same ligand, these two integrins have distinct and specific roles in cell migration [13].
Adhesion to fibronectin by αvβ3 integrin supports extensive actin cytoskeletal reorganization resulting in a single large lamellipod with static cell–matrix adhesions at the leading edge [14]. On the other hand, cell adhesion by α5β1 generates thin protrusions containing highly dynamic cell–matrix adhesions in multiple directions [14]. Therefore, these authors concluded that β1 integrins support random migration, whereas β3 integrins are related to persistent migration. In agreement with these data, we have demonstrated that blockage of αvβ3 integrin by a DisBa-01, a recombinant RGD-disintegrin from snake venom, resulted in loss of directionality and decrease of speed migration [15]. Despite having the RGD adhesive motif, which is recognized by both αvβ3 and α5β1 integrins, the dissociation constant of DisBa-01 for the αvβ3 integrin is 100 times higher than for α5β1 integrin [15]. These results confirm the role of αvβ3 integrin in defining directionality of the cell movement.
Rosa-Cusachs et al., 2009 also provided evidence of distinct roles for α5β1 and αvβ3 integrin during cell adhesion/migration processes, with the demonstration that α5β1 integrin is responsible for supporting high adhesion forces while αvβ3 integrin starts talin-dependent mechanotransduction. However, these effects depend on the substrate where cells attach. Fibroblasts exhibit persistence migration on FN-coated surfaces [16]. Ligand-specific activation of αvβ3 e α5β1 integrins also confirmed the distinct roles of each integrin in cell adhesion. Activation of αvβ3 integrins led to stabilization of peripheral focal adhesions while fibrillary structures were observed when α5β1 integrins are activated [17]. Moreover, αv-class of integrins such as αvβ3 were demonstrated to outcompete α5β1 integrins in FN binding; however, after engagement, αv-class integrins cooperate with α5β1 integrins to form additional adhesion sites consequently strengthening cell adhesion to FN [18].
Although they may bind the same ligand, β1 and β3 integrins have distinct and cooperative roles in mechanotransduction. By means of traction force microscopy, Millioud and colleagues demonstrated that deleting β3 subunit increases traction forces, whereas the deletion of β1 subunit results in a strong decrease of contractile forces [19].
Integrins and Growth Factor Receptors
The cross talk between integrin and growth factor receptor (GFR) signaling pathways is well documented in the literature in both normal cells and tumor cells. Endothelial cells seem to be highly sensitive to integrin activation of GFRs including the vascular endothelial growth factor receptor (VEGFR), epidermal growth factor receptor (EGFR), and the platelet-derived growth factor receptor (PDGFR). This cooperative process is crucial in physiological and tumor angiogenesis and it is considered one key factor in tumor progression. The α5β1 integrin was demonstrated to induce tumor cell proliferation by two pro-survival mechanisms including direct activation the EGFR signaling cascade and by signaling via continuous integrin-dependent activation of protein kinase B (PKB, also known as AKT) [20]. Moreover, EGFR overexpression led to inactivation of α5β1 integrin in A431 squamous carcinoma cells, which would be interesting in order to prevent cell interaction with the ECM and therefore to avoid tumor cell proliferation. However, treatment of cells with EGFR kinase inhibitor resulted in reactivation of the integrin [21]. Since GFRs are usually overexpressed in many types of cancer cells, pro-survival signaling pathways from both GFR and integrins would be more efficiently impaired upon association of anti-integrin/anti-GFR treatments than either treatment alone [22].
VEGF binding to its receptors and co-receptors induce receptor homodimerization and heterodimerization, followed by activation of tyrosine kinases and signaling cascades, with the association of a set of adaptor proteins. The activation of these signaling pathways results in activation, migration and proliferation of endothelial cells, crucial steps for neoangiogenesis. In addition, mechanical forces such as shear stress may activate VEGRF2 similarly to ligand binding, leading to the activation of intracellular signaling pathways [23]. Integrins are proposed to act as co-receptors upon VEGF binding to VEGFRs, similarly to neuropilins and heparin sulfate proteoglycans in endothelial cells [24]. However, integrins seem to participate actively in the control of VEGFR signaling. Blocking αvβ3 integrin by a RGD-antagonist downregulated the expression of VEGF, VEGFR1 and VEGFR-2 in endothelial cells but not in MDA-MB-231 breast tumor cells [25]. Contrastingly, we observed an increase in VEGF protein levels by human fibroblasts after treatment with the αvβ3 integrin antagonist. In addition, we have also demonstrated that this RGD-antagonist inhibits endothelial in vitro cell migration and in vivo angiogenesis in mice [26, 27]. These results suggest that the αvβ3 integrin is not only a co-receptor; instead, it is an active partner in the control of VEGF signaling in endothelial cells.
A key role for α1β1 and α2β1 integrins in the process of angiogenesis triggered by vascular endothelial growth factor (VEGF) was previously reported. Antibody antagonism of either integrin resulted in potent inhibition of VEGF-driven angiogenesis in mouse skin, reduced tumor growth and angiogenesis of human squamous cell carcinoma xenografts. [28]
Integrins and MMPs
Matrix metalloproteases (MMP) comprise a class of zinc-dependent enzymes responsible for ECM remodeling and degradation. MMPs are also involved in the processing of growth factors, cytokines and surface transmembrane proteins. MMPs are produced as inactive zymogens that can be activated by proteolytic removal of the pro-peptide domain by furin, or by autoproteolysis. To date, more than 24 MMPs are known, including secreted or membrane anchored forms, (membrane-type MMPs, MT-MMPs) that play crucial roles in the process of MMP activation. For instance, activation of pro-MMP-2 at the cell surface involves the formation of a trimolecular complex with membrane type-1 metalloprotease (MT-1-MMP) and tissue inhibitor of metalloproteases-2 (TIMP-2). MMP-2 and MMP-9 are also known as gelatinases A and B, respectively, due to the ability to digest degraded forms of collagen. Gelatinases are characterized by the presence of a fibronectin-like domain that drives the enzyme to its ECM substrates. MMP-2 and MMP-9 are of particular interest in cell migration and consequently, in metastasis development. MMP-2 is constitutively produced by most cells; however, MMP-9 is expressed by only a few types of cells including neutrophils, macrophages and tumor cells, being induced in several pathological conditions such as tumor invasion [29, 30].
MMPs play key roles in tumor progression such as degrading ECM to allow migration of tumor cells to distant secondary sites, allowing endothelial cell migration and proliferation to produce tumor angiogenesis, and releasing growth factors from the ECM to provide constant survival and proliferation signals.
The role of integrins in controlling MMP activity is less studied. Previous studies demonstrated that interaction of fibronectin with α4β1 integrin upregulates MMP-9 expression by infiltrating leukocytes in damaged livers and the blockade of this interaction disrupted leukocyte migration [30]. MMP-2 activation upregulates VEGF-A expression in melanoma cells via an αvβ5 integrin/phosphoinositide-3-kinase-dependent pathway [31]. The αvβ3 integrin has been closely related to tumor progression and reduced patient survival rates in melanoma, colon carcinoma and breast cancer, increasing migration and invasion of tumor cells [32, 33]. Blocking αvβ3 integrin inhibited MT-1-MMP-dependent activation of MMP-2 induced by collagen I; however, cells expressing high levels of β3 integrin subunit have increased abilities of adhesion and migration [34]. Blocking αvβ3 integrin in endothelial cells by a RGD-based antagonist completely abolished MMP-2 activity; in contrast, the same treatment increased almost twice the MMP-9 levels in the conditioned media from MDA-MB-231 breast tumor cells [25]. These results demonstrated that one single specific integrin inhibitor might induce different cell-dependent effects.
Integrins and tumor progression
The correlation of expression levels of αvβ3, αvβ5, α5β1, α6β4, α4β1, αvβ6 and αvβ8 integrins with metastasis and poor patient prognosis is well documented (reviewed by Nieberler et al., 2017). [35] One of the most studied integrin in tumor progression is the α5β1 integrin. Higher levels of α5 subunit were found than in normal tissue in patients with advanced renal cell carcinoma and correlated with tumor grade, metastasis development and reduced patient survival [36]. Expression of αvβ6 is significantly associated with the progression of breast ductal carcinoma to an invasive form by a mechanism involving upregulation of MMP-9 and transforming growth factor-β (TGF-β) [37].
Previous reports associated β1 and β3 integrins with TGF-β stimulation of epithelial–mesenchymal transition (EMT) and breast tumor metastasis by means of a compensatory mechanism [38]. Inactivation of β1 integrin impairs the TGF-β effect in promoting tumor cell migration; however, a strong compensatory upregulation of β3 integrin restores the induction of the EMT phenotypes by TGF-β, indicating that the two integrins must be targeted to prevent tumor progression [38].
Integrin-targeted therapy
Cilengitide was one of the first antiangiogenic drugs directed to blockade of cell adhesion to the ECM by antagonizing αvβ3 and α5β1 integrins (IC50 for inhibition of cell adhesion of 0.2 and 11 nM, respectively). In phase I studies, patients with advanced solid tumors were treated with cilengitide without consistent results [39]. In another study, cilengitide was tested in association with cediranib, an inhibitor of VEGFR-associated tyrosine kinase. The association of cilengitide with the two drugs was well tolerated, however there were no changes in the survival rates [40, 41]. Cilengitide combined with temozolomide, an oral alkylating agent, did not increase the survival rates of glioblastoma patients [42]. Among antiangiogenic drugs, only the anti-VEGF-A bevacizumab increased disease-free survival time in patients with glioblastoma [43]. Recently, 12 patients with solid tumors such as breast cancer were treated with cilengitide, with partial positive response to the treatment and 05 had stable disease as the best response [44]. In summary, after 10 years of clinical assays with cilengitide for different tumors, results are still disappointing. Reasons for the lack of success may be related to the determination of the effective doses and time of administration. [45- 47] Other reason for lack of success may be the dose-dependent opposing effects of cilengitide related to tumor angiogenesis, with low doses being pro-angiogenic in contrast with anti-angiogenic effect of higher doses [48]. However, a better comprehension on the distinct roles of integrins in the context of complexity of the tumor microenvironment is needed before discarding integrin-targeted therapy.
Volociximab, a monoclonal antibody anti-α5β1 integrin, has been tested in at least 10 phase I and II clinical trials to treat some types of tumors, including advanced non-small cell lung cancer (NSCLC) and metastatic melanoma, among others. Volociximab was used either as single therapy or in combination with classical drugs such as carboplatin and paclitaxel. Preliminary results showed modest but relevant results such as an increase in median progression-free survival of 6.3 months and decreased levels of potential biomarkers of angiogenesis or metastasis after six cycles of treatment [49].
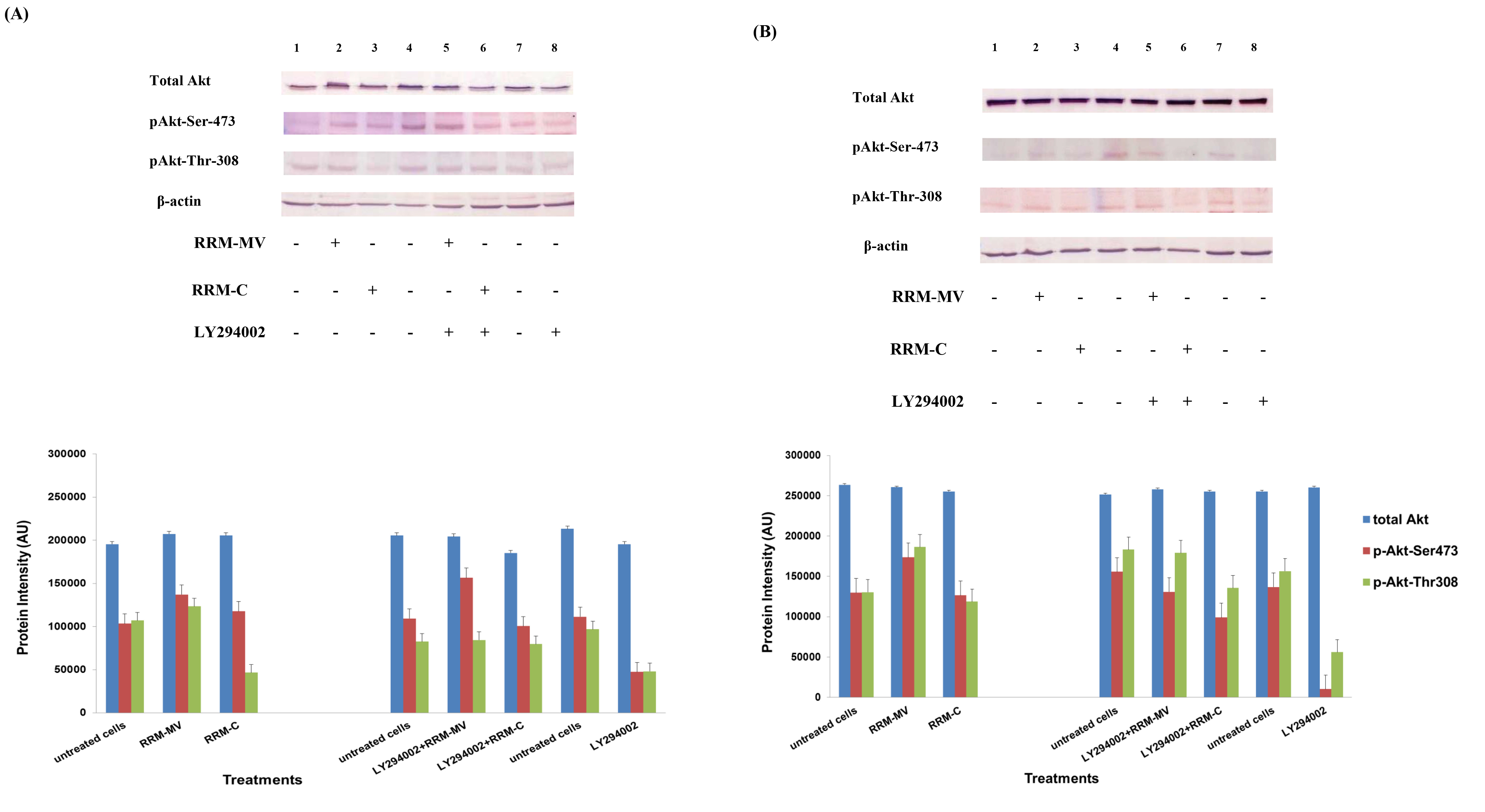
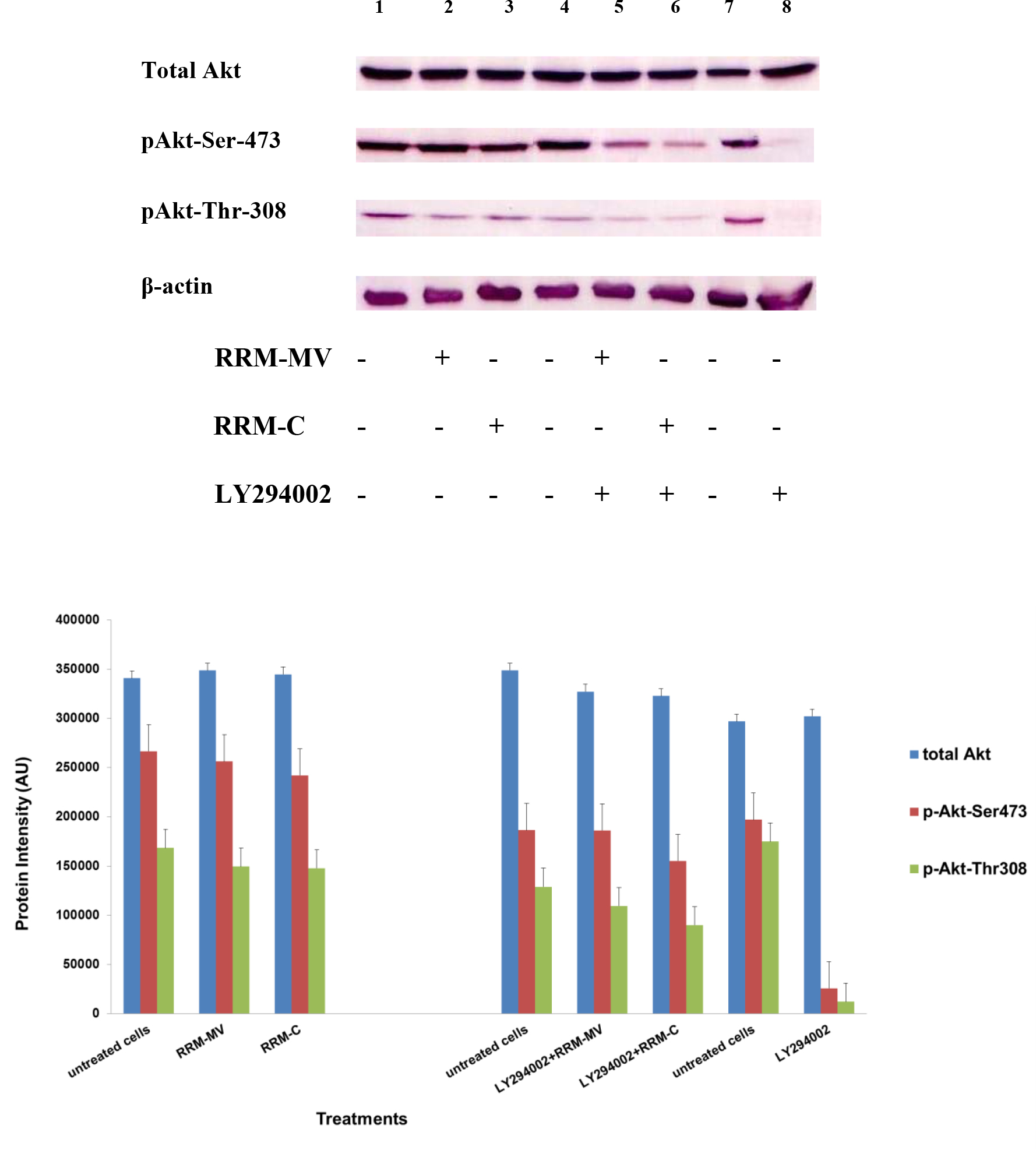
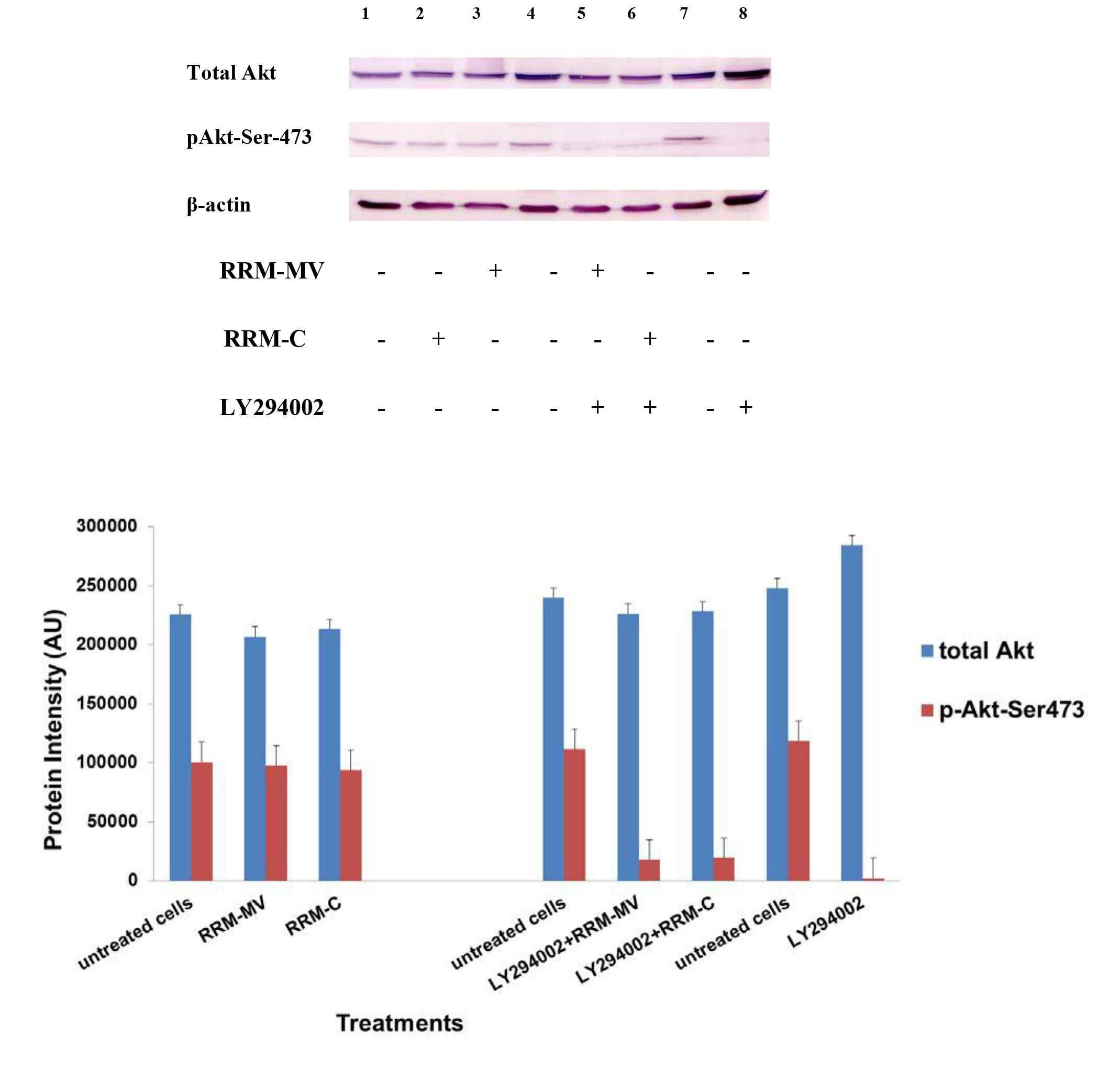
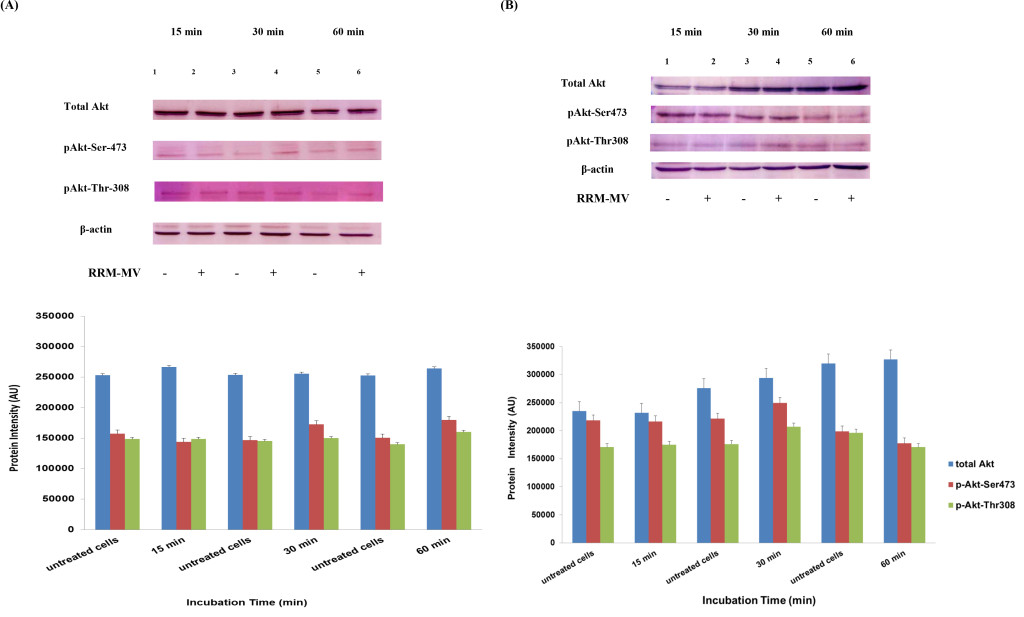
Etaracizumab, an anti-αvβ3 integrin monoclonal antibody, decreased SKOV3ip1 ovarian tumor cell proliferation and invasion in vitro and resulted in about 50% of tumor weight decrease in mice [50]. Combination therapy with paclitaxel gave better results in decreasing tumor weight, and tumors showed reduced levels of p-Akt and p-mTOR; however, microvessel density of resected tumors after therapy were not decreased [50]. The β1 integrin subunit has been associated to therapeutic resistance to trastuzumab (anti-EGFR/HER2 monoclonal antibody) and to lapatinib (an EGFR/HER2 tyrosine kinase inhibitor) of human epidermal growth factor receptor (HER)-2-positive breast tumor cells [51].
One of the most intriguing question in integrin-based anti-cancer therapy is the fact that the strong and positive results on inhibition of tumor progression in pre-clinical assays did not translate to clinical assays. Integrin inhibitors including monoclonal antibodies and synthetic molecules showed disappointed results in patient survival time, disease stabilization and the development of metastasis [42, 48, 52]. One reason for the negative results may rely on the complexity of the mechanism of action of integrins, their ability to compensate each other and inducing an even worse phenotype. Deleting β1 integrin was compensated by β3 integrin, which stimulated metastasis in murine model of breast cancer [38].
Besides the compensatory mechanism, integrin inhibition should induce cell death by anoikis, a kind of apoptotic cell death that occurs due to the loss of cell attachment to the ECM [53]. However, ECM detachment results in antiapoptotic signals, as a defense mechanism against anoikis until cells be able to find a new place to attach again. Meantime, cells undergo autophagy, a cell process mostly triggered by the loss of integrin-mediated adhesion that allow cell survival during some time. However, prolonged detachment will later induce apoptosis by anoikis [54]. One of the most critical finding is that tumor cells usually develop resistance to anoikis due to a sustained autophagic response [54].
Entosis, an even more sophisticated cell survival mechanism, was described [55]. Entosis, also referred to as cell-in cell structures, or cell cannibalism, is triggered by loss of attachment to ECM, similarly to the process of autophagy. Entotic cells have been observed in many types of tumors and may be one of the reasons for the abnormal number of chromosomes found in most tumors [2]. Engulfed cells are alive and may divide inside the host, and in case of tumor cells, such process may occur indefinitely. Such mechanisms of tumor cell survival that happen upon ECM detachment may certainly contribute for the lack of success of integrin inhibitors in clinical trials.
More recently, the mechanism of vessel co-option was described as a mediator of resistance to anti-angiogenic therapy of breast tumor liver and lung metastasis [56]. Vessel co-option is an alternative pathway of tumor cells for obtaining nutrients from blood using the pre-existing vasculature without producing new vessels. Blocking VEGF/VEGFR signaling induces co-option and tumor growth in glioblastoma patients. [57] Since there is a close reciprocal stimulatory relationship between VEGF and αvβ3 integrin [58], one might expect a role for integrin inhibition in supporting the mechanism of co-option. This possibility remains to be elucidated. These results demonstrate the complexity of the TME and its relevance in tumor progression.
New insights on integrin targeting
In spite of being extensively described, integrin inhibition in tumor microenvironment is still challenging and attractive. The failure of targeted therapies so far leads to deeper investigations about signaling pathways, endocytic trafficking and recycling of integrins [59, 60].
As a result, the attention that before was outside cell, turned to intracellular integrin fate affecting the overall cell behavior. Its known that integrin trafficking dictates the nature of Rho GTPase signaling during cytokinesis and cell migration [61]. FN binding promotes both accelerated internalization and ubiquitination of α5β1 receptors. Subsequently, alterations in pH inside endossomal compartments will define either recycling or degradation of integrins [59, 62] When α5 integrin suffers ubiquitination upon FN binding, the ESCRT machinery acts sorting this receptor to either multivesicular bodies, recycling or lysosomal degradation [59]. It was previously demonstrated that mutation on the ubiquitination site in cytoplasmic tail of α5 integrin causes a different sorting of fibronectin on cell, affecting fibroblast migration [63].
The multivesicular bodies are cell compartments containing intraluminal vesicles named exosomes that are secreted to the extracellular space by shedding from the plasma membrane, improving cell communication with the TME [64]. The presence of both integrin and FN inside multivesicular bodies had light to the hypothesis that these receptors might be present in vesicles in the extracellular environment. Sung et al demonstrated that in fact α5 integrin is secreted as an exosome cargo, and more than that, FN was also secreted in these vesicles. Thus, a new and promising science of integrins as a target has emerged from the microvesicles field. Studies on cell communication had proved that integrin transfer can occur through exosomes delivery in both TME and the pre-metastatic niche [65]. In addition, the integrin content of exosomes can change the types of integrin expression in the new tumor focus [66]. The real contribution of integrins on cell communication using cell-derived vesicles is still unclear, as well as the effects of blockage of these receptors. However, this can be the missing clue that can explain the failure of earlier integrin inhibitors in drug development.
Conclusions
Cell attachment to the ECM via integrins is a key factor for tumor progress; however, integrin inhibition may be carefully considered as a target for drug development. Combination of anti-growth factors antibodies, tyrosine kinase inhibitors and integrin inhibitors may be an interesting choice but must be first evaluated in pre-clinical assays considering all the possible escape mechanisms that tumor cells can develop. Most of the studies so far have considered the signaling in a cellular level, however, in complex organisms, several other factors might systemically interfere on integrin inhibition, making the drug development even more challenging. The integrins may have won some battles but not the war.
Acknowledgements: This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, 2013/00798-2 and 2014/18747-8) and by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq 308096/2013-4), Brazil.
References
- Steeg PS (2016) Targeting metastasis. Nat Publ Gr 16.
- Janssen A, Medema RH (2011) Entosis: aneuploidy by invasion. Nat Cell Biol 13: 199–201.
- Desgrosellier JS, Cheresh DA (2010) Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 10: 9–22.
- Brücher BLDM, Jamall IS (2014). Cell-Cell Communication in the Tumor Microenvironment, Carcinogenesis, and Anticancer Treatment. Cell Physiol Biochem 34: 213–43.
- Korkaya H, Liu S, Wicha MS (2011) Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest 121: 3804–9.
- Wu T, Dai Y (2017) Tumor microenvironment and therapeutic response. Cancer Lett 387: 61–8.
- Longmate W, DiPersio CM (2017) Beyond adhesion: emerging roles for integrins in control of the tumor microenvironment. F1000Research 6: 1612.
- Hynes RO (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110: 673–87.
- Hynes RO (1987) Integrins: a family of cell surface receptors. Cell 48: 549–554. [crossref]
- Nieberler M, Reuning U, Reichart F, Notni J, Wester H-J, et al. (2017) Exploring the Role of RGD-Recognizing Integrins in Cancer. Cancers (Basel) 9: 116.
- Montenegro CF, Casali BC, Lino RLB, Pachane BC, Santos PK, et al. (2017) Inhibition of avß3 integrin induces loss of cell directionality of oral squamous carcinoma cells (OSCC). Languino LR, editor. PLoS One 12: e0176226.
- Miranti CK, Brugge JS (2002) Sensing the environment: a historical perspective on integrin signal transduction Early studies on the regulation of cell behaviour by adhesion. Nat Cell Biol
- Roca-Cusachs P, Gauthier NC, del Rio A, Sheetz MP (2009) Clustering of 5 1 integrins determines adhesion strength whereas v 3 and talin enable mechanotransduction. Proc Natl Acad Sci 106: 16245–50.
- Danen EHJ, van Rheenen J, Franken W, Huveneers S, Sonneveld P, et al. (2005) Integrins control motile strategy through a Rho–cofilin pathway. J Cell Biol 169: 515–26.
- Montenegro CF, Casali BC, Lino RLB, Pachane BC, Santos PK, et al. (2017) Inhibition of avß3 integrin induces loss of cell directionality of oral squamous carcinoma cells (OSCC). Languino LR, editor. PLoS One 12: e0176226.
- Missirlis D, Haraszti T, Scheele C v. C, Wiegand T, Diaz C, et al. (2016) Substrate engagement of integrins a5ß1 and avß3 is necessary, but not sufficient, for high directional persistence in migration on fibronectin. Sci Rep 6: 23258.
- Schaufler V, Czichos-Medda H, Hirschfeld-Warnecken V, Neubauer S, Rechenmacher F, et al. (2016) Selective binding and lateral clustering of a 5 ß 1 and a v ß 3 integrins: Unraveling the spatial requirements for cell spreading and focal adhesion assembly. Cell Adh Migr 10: 505–15.
- Bharadwaj M, Strohmeyer N, Colo GP, Helenius J, Beerenwinkel N, et al. (2017) aV-class integrins exert dual roles on a5ß1 integrins to strengthen adhesion to fibronectin. Nat Commun.
- Milloud R, Destaing O, de Mets R, Bourrin-Reynard I, Oddou C, et al. (2017) avß3 integrins negatively regulate cellular forces by phosphorylation of its distal NPXY site. Biol Cell 109: 127–37.
- Morozevich GE, Kozlova NI, Ushakova NA, Preobrazhenskaya ME, Berman AE (2012) Integrin a5ß1 simultaneously controls EGFR-dependent proliferation and Akt-dependent pro-survival signaling in epidermoid carcinoma cells. Aging (Albany NY) 4: 368–74.
- Vial D, McKeown-Longo PJ (2016) Role of EGFR expression levels in the regulation of integrin function by EGF. Mol Carcinog 55: 1118–23.
- Eke I, Zscheppang K, Dickreuter E, Hickmann L, Mazzeo E, et al. (2015) Simultaneous β1 integrin-EGFR targeting and radiosensitization of human head and neck cancer. J Natl Cancer Inst 107. [crossref]
- Jin Z-G, Ueba H, Tanimoto T, Lungu AO, Frame MD, Berk BC (2003) Ligand-Independent Activation of Vascular Endothelial Growth Factor Receptor 2 by Fluid Shear Stress Regulates Activation of Endothelial Nitric Oxide Synthase. Circ Res 93: 354–63.
- Simons M, Gordon E, Claesson-Welsh L (2016) Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol 17: 611–25.
- Montenegro CF, Salla-Pontes CL, Ribeiro JU, Machado AZ, Ramos RF, et al. (2012) Blocking avß3 integrin by a recombinant RGD disintegrin impairs VEGF signaling in endothelial cells. Biochimie. 94: 1812–20.
- Ramos OHP, Kauskot A, Cominetti MR, Bechyne I, Salla Pontes CL, et al. (2008) A novel alpha(v)beta (3)-blocking disintegrin containing the RGD motive, DisBa-01, inhibits bFGF-induced angiogenesis and melanoma metastasis. Clin Exp Metastasis 25: 53–64.
- Cassini-Vieira P, Deconte SR amos, Tomiosso TC arla, Campos PP eixoto, Montenegro C de F, et al. (2014) DisBa-01 inhibits angiogenesis, inflammation and fibrogenesis of sponge-induced-fibrovascular tissue in mice 92: 81–9.
- Senger DR, Perruzzi CA, Streit M, Koteliansky VE, de Fougerolles AR, Detmar M (2002) The alpha(1)beta(1) and alpha(2)beta(1) integrins provide critical support for vascular endothelial growth factor signaling, endothelial cell migration, and tumor angiogenesis. Am J Pathol 160: 195–204.
- Egeblad M, Werb Z (2002) New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2: 161–174. [crossref]
- Hamada T, Fondevila C, Busuttil RW, Coito AJ (2007) Metalloproteinase-9 deficiency protects against hepatic ischemia/reperfusion injury. Hepatology 47: 186–98.
- Desch A, Strozyk EA, Bauer AT, Huck V, Niemeyer V, et al. (2012) Highly Invasive Melanoma Cells Activate the Vascular Endothelium via an MMP-2/Integrin avß5–Induced Secretion of VEGF-A. Am J Pathol 181: 693–705.
- Cooper CR, Chay CH, Pienta KJ (2002) The role of alpha(v)beta(3) in prostate cancer progression. Neoplasia. 4: 191–4.
- Pécheur I, Peyruchaud O, Serre C-M, Guglielmi J, Voland C, et al. (2002) Integrin avß3 expression confers on tumor cells a greater propensity to metastasize to bone. FASEB J 16: 1266–8.
- Borrirukwanit K, Lafleur MA, Mercuri FA, Blick T, Price JT, et al. (2007) The type I collagen induction of MT1-MMP-mediated MMP-2 activation is repressed by aVß3 integrin in human breast cancer cells. Matrix Biol 26: 291–305.
- Nieberler M, Reuning U, Reichart F, Notni J, Wester H-J, et al. (2017) Exploring the Role of RGD-Recognizing Integrins in Cancer. Cancers (Basel) 9: 116.
- Breuksch I, Prosinger F, Baehr F, Engelhardt F-P, Bauer H-K, et al. (2017) Integrin a5 triggers the metastatic potential in renal cell carcinoma. Oncotarget 8: 107530–42.
- Allen MD, Thomas GJ, Clark S, Dawoud MM, Vallath S, et al. (2014) Altered Microenvironment Promotes Progression of Preinvasive Breast Cancer: Myoepithelial Expression of v 6 Integrin in DCIS Identifies High-risk Patients and Predicts Recurrence. Clin Cancer Res 20: 344–57.
- Parvani JG, Galliher-Beckley AJ, Schiemann BJ, Schiemann WP (2013) Targeted inactivation of 1 integrin induces 3 integrin switching, which drives breast cancer metastasis by TGF-. Mol Biol Cell. 24: 3449–59.
- Hariharan S, Gustafson D, Holden S, McConkey D, Davis D, et al. (2007) Assessment of the biological and pharmacological effects of the a?ß3 and a?ß5 integrin receptor antagonist, cilengitide (EMD 121974), in patients with advanced solid tumors. Ann Oncol 18: 1400–7.
- Friess H, Langrehr JM, Oettle H, Raedle J, Niedergethmann M, et al. (2006) A randomized multi-center phase II trial of the angiogenesis inhibitor Cilengitide (EMD 121974) and gemcitabine compared with gemcitabine alone in advanced unresectable pancreatic cancer. BMC Cancer 6: 285
- Gerstner ER, Ye X, Duda DG, Levine MA, Mikkelsen T, et al. (2015) A phase I study of cediranib in combination with cilengitide in patients with recurrent glioblastoma. Neuro Oncol 17: 1386–92.
- Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, et al. (2014) Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 15: 1100–8.
- Lombardi G, Pambuku A, Bellu L, Farina M, Della Puppa A, et al. (2017) Effectiveness of antiangiogenic drugs in glioblastoma patients: A systematic review and meta-analysis of randomized clinical trials. Crit Rev Oncol Hematol 111: 94–102.
- Haddad T, Qin R, Lupu R, Satele D, Eadens M, et al. (2017) A phase I study of cilengitide and paclitaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol 79: 1221–7.
- Steri V, Ellison TS, Gontarczyk AM, Weilbaecher K, Schneider JG, et al. (2014) Acute depletion of endothelial ß3-integrin transiently inhibits tumor growth and angiogenesis in mice. Circ Res 114: 79–91.
- Atkinson SJ, Ellison TS, Steri V, Gould E, Robinson SD (2014) Redefining the role(s) of endothelial avß3-integrin in angiogenesis: Figure 1. Biochem Soc Trans 42: 1590–5.
- Blandin A-F, Renner G, Lehmann M, Lelong-Rebel I, Martin S, Dontenwill M (2015) ß1 Integrins as Therapeutic Targets to Disrupt Hallmarks of Cancer. Front Pharmacol 6: 279.
- Chinot OL1 (2014) Cilengitide in glioblastoma: when did it fail? Lancet Oncol 15: 1044–1045. [crossref]
- Besse B, Tsao LC, Chao DT, Fang Y, Soria J-C, et al. (2013) Phase Ib safety and pharmacokinetic study of volociximab, an anti-a5ß1 integrin antibody, in combination with carboplatin and paclitaxel in advanced non-small-cell lung cancer. Ann Oncol 24: 90–6.
- Landen CN, Kim T-J, Lin YG, Merritt WM, Kamat AA, et al. (2008) Tumor-selective response to antibody-mediated targeting of alphavbeta3 integrin in ovarian cancer. Neoplasia 10: 1259–67.
- Huang C, Park CC, Hilsenbeck SG, Ward R, Rimawi MF, et al. (2011) ß1 integrin mediates an alternative survival pathway in breast cancer cells resistant to lapatinib. Breast Cancer Res 13: R84.
- Kobayashi M, Sawada K, Kimura T (2017) Potential of integrin inhibitors for treating ovarian cancer: A literature review. Cancers (Basel).
- Frisch SM, Ruoslahti E (1997) Integrins and anoikis. Curr Opin Cell Biol 9: 701-706. [crossref]
- Vlahakis A, Debnath J (2017) The Interconnections between Autophagy and Integrin-Mediated Cell Adhesion. J Mol Biol 429: 515–30.
- Overholtzer M, Mailleux AA, Mouneimne G, Normand G, Schnitt SJ, et al. (2007) A Nonapoptotic Cell Death Process, Entosis, that Occurs by Cell-in-Cell Invasion. Cell 131: 966–79.
- Frentzas S, Simoneau E, Bridgeman VL, Vermeulen PB, Foo S, et al. (2016) Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat Med 22: 1294–302.
- Rubenstein JL, Kim J, Ozawa T, Zhang M, Westphal M, et al. (2000) Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia 2: 306–14.
- Brakenhielm E (2007) Substrate Matters: Reciprocally Stimulatory Integrin and VEGF Signaling in Endothelial Cells. Circ Res 101: 536–8.
- Lobert VH, Stenmark H (2010) Ubiquitination of a-integrin cytoplasmic tails. Commun Integr Biol. 3: 583–5.
- Paul NR, Jacquemet G, Caswell PT. Endocytic Trafficking of Integrins in Cell Migration. Curr Biol 25: R1092–105.
- Caswell PT, Vadrevu S, Norman JC. Integrins: masters and slaves of endocytic transport. Nat Rev Mol Cell Biol. 10: 843–53.
- Kharitidi D, Apaja PM, Manteghi S, Suzuki K, Malitskaya E, et al. (2015) Interplay of Endosomal pH and Ligand Occupancy in Integrin?a5b1 Ubiquitination, Endocytic Sorting, and Cell Migration. Cell Rep 13: 599–609.
- Brech A, Pedersen NM, Wesche J, Oppelt A (2010) Article Ubiquitination of a 5 b 1 Integrin Controls Fibroblast Migration through Lysosomal Degradation of Fibronectin-Integrin Complexes 148–59.
- Raposo G, Stoorvogel W (2013) Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 200: 373–383. [crossref]
- Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, et al. (2015) Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol 17: 816–26.
- Hoshino A, Costa-Silva B, Shen T-L, Rodrigues G, Hashimoto A, et al. (2015) Tumour exosome integrins determine organotropic metastasis. Nature 527: 329–35.