DOI: 10.31038/IJVB.2018221
Abstract
Cooled and frozen equine semen shows a reduction in fertility, compared to fresh one. In this study, cooled and frozen-thawed equine spermatozoa were compared and analyzed for plasma and acrosomal membrane integrity and mitochondrial membrane potential, combining three fluorescent probes: H258, CTC, JC-1 with a micro-spectrofluorimetric analysis (Quanticell equipped with a digital system for color images acquisition). Total and progressive motility, average path velocity (VAP), straight-line velocity (VSL), curvilinear velocity (VCL) and amplitude of lateral head displacement (ALH) were measured by CASA system.
We employed an innovative approach to study the reproductive potential of the male gamete subjected to cooling protocols for semen storage. In fact, we evaluated the modifications of equine sperm physiology induced by temperature during cooling and freezing treatments looking at the modifications of different functional sperm characteristics by a simultaneous analysis of different sperm markers with the aim of selecting those that are the most efficient signs for sperm fertility. We identified the mitochondrial membrane potential because it provides useful information on equine sperm quality strictly correlated with fertility. We consider it a useful marker for sperm fertility to be used as a guide to select high-quality semen to be employed in equine breeding farmers.
Keywords
Stallion; Sperm; Cooled semen; frozen semen; Sub-lethal damages
Introduction
Nowadays reproductive technologies are more attractive for the equine industry [1] because of the increasing use of both refrigerated and frozen sexed spermatozoa. Despite the high number of published papers, further knowledge on stallion sperm cryobiology still requires investigations. In the equine species, sperm quality is affected by inter-individual variability, because stallion selection is often based on performance and phenotype more than on sperm quality [2].
Short- and long-term sperm storage is a pre-requisite for the success of artificial insemination. Cooled-stored stallion semen is usually kept at a temperature of about 4-6°C for about 24h. This range of temperature has been evaluated to be efficacious for maintaining motility and consequently fertility near to fresh semen [3] Long-term storage is useful to preserve gametes from high merit animals, to check the health status of semen samples and to share superior genetics among international distributors [4]. Survival of the sperm cell to refrigeration and cryopreservation depends on its shape and size, on its hydration level and on the permeability of its cell membrane. Refrigerated and frozen-thawed semen, used in different artificial insemination programs, show some limits regards viability compared to fresh semen, due to changes in temperature. Temperature variations during a rapid cooling or during cryopreservation of spermatozoa are in fact known to exert deleterious effects on the survival of spermatozoa, resulting in lower conception rates following artificial insemination. Fertility decrease is the result of the reduction of the percentage of motile sperm, or the consequence of morphological abnormalities induced by cryopreservation, as well as of damages of sperm cell plasma membrane [5], that are responsible of the induction of cell death and sub-lethal damages in the surviving population of spermatozoa [6]. Semen evaluation represents a useful tool to investigate male infertility [7] and in many circumstances, a prospective test is desirable to identify an infertile stallion before it embarks on its breeding career [8]. In horse breeding, fertility trials associated to artificial insemination techniques are hampered by the low number of fertilized mares and by the high difficulties in management and insemination [9]. Given the limitations of the standard examination procedures in predicting horse fertility or even in identifying all infertile stallions, many approaches were employed with the hope of defining a relatively straightforward and inexpensive test closely correlated with fertility [10]. The limitation of these approaches is that most tests evaluate only a limited number of those characteristics necessary for the assessment of sperm fertility rate. For this reason, the combined use of different tests could be more promising to achieve a reliable evaluation of the functional characteristics related to sperm quality [8, 11, 12].
The integrity of sperm membrane is crucial for the maintenance of sperm fertilizing capacity [7] in fact, it is a fundamental requisite for sperm viability and for the success of fertilization. Viable spermatozoa are defined as cells that possess an intact plasma membrane [9]. Several viability assays evaluate the integrity of different plasma membrane compartments by a microscopic or cytofluorimetric approach after cell staining [9, 13]. These techniques, based on the use of viable (SYBR-14) and non-viable propidium iodide (PI) and Hoechst 33258 (H258) dyes allow detecting sperm viability [14].
Acrosome integrity is essential for oocyte fertilization [7, 15], different fluorescent staining such as Pisum sativum agglutinin (PSA) and chlortetracycline (CTC) perform its evaluation. Besides plasma membrane and acrosome integrity, the mitochondrial status plays an important role in determining sperm cell fertility competence because of its relationship with the energetic status of the cell and with motility [9]. The functionality of mitochondria is often studied by rhodamine 123 and MitoTracker fluorochromes but these techniques do not possess the ability to differentiate mitochondria with low or high membrane potential [16]. The lipophilic cationic compound, 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethylbenzimidazolyl carbocyanine iodide (JC-1) allows for a distinction between spermatozoa with low and high functional mitochondria since this molecule possesses the ability to form aggregates or monomers, each endowed with a different emission spectrum. JC-1 is maintained as a monomer in mitochondria with low membrane potential, emitting green fluorescence, whereas it aggregates in mitochondria with high membrane potential, emitting orange fluorescence. Several authors still employ the sperm chromatin structure assay test (SCSA), based on the metachromatic properties of the acridine orange, to evaluate breaks in the DNA that may have escaped repair during the last steps of spermatogenesis.
Sperm motility is a very important parameter for sperm quality evaluation and it can be assessed using Computer Assisted Sperm Analysis (CASA) system that captures and digitized successive microscopic images [9, 17].
Fluorescent probe association is able to perform a simultaneous evaluation of several sperm cell compartments [18]. It is intuitive that the higher the number of sperm characteristics, the better is the in vitro fertility prognosis [19].
The aim of this study is to evaluate damages, induced by temperature during refrigeration and cryopreservation, on the plasma, acrosomal and mitochondrial membrane of equine sperm cells and to correlate them in order to select the best characteristics useful for a reliable prediction of the fertilizing competence of each semen sample employed in equine breeding.
Materials and methods
Ethical statement
The experiment was approved by the Ethical and Scientific Committee of “Alma Mater Studiorum”, University of Bologna”
Media and Reagents
A modified Tyrode’s medium without bicarbonate was used for incubating sperm in ‘‘non-capacitating’’ conditions. The Tyrode’s medium contained 111 mM NaCl, 3.1 mM KCl, 2 mM CaCl2, 0.4 mM MgSO4, 0.3 mM KH2PO4, 50 mg kanamycin/ml, 20 mM HEPES, 5 mM glucose, 21.7 mM sodium lactate, 1 mM sodium pyruvate. Stock solutions minus CaCl2 and pyruvate were prepared, filtered through a 0.2 mm membrane and stored at +4°C [20]. The remaining two ingredients were added 20-24 h before the experiment and the medium maintained under 5% CO2 in air at +37°C until the beginning of the experiment; pH and osmolality of the medium were maintained at 7.4 and 300 mOsm/kg, respectively.
All chemicals were purchased from Sigma-Aldrich (Milano, Italy) unless otherwise stated.
Semen collection and processing
Semen (eight ejaculates per stallion) was obtained from two individually housed stallions (Standarbred, 6 and 7 years old), routinely used in the artificial insemination programs at AUB-National Institute for Artificial Insemination, located in Cadriano (Bologna, Italy). The management of the stallions and the collection of semen samples were performed in accordance with health and welfare institutional and European regulations.
Ejaculates were collected regularly (three times/week) during the breeding season of 2011, using a Missouri artificial vagina with an in-line filter (Nasco, Fort Atkinson, WI, USA). Gel-free semen volume was measured with a graduated cylinder, semen concentration was determined using a Bürker chamber (Saaringia, Germany) and motion characteristics were estimated with CASA system. Subsequently, the filtered semen was diluted with Kenney’s extender [21] supplemented with a Tyrode medium, to a final concentration of about 20-25 × 106 spermatozoa ⁄ml. One ml aliquot of each fresh semen sample was evaluated by CASA system. After a 10 min incubation period at +37°C, 2 µl of the suspension was loaded onto a pre-warmed (37°C) Leja 20 µm four chamber slide (IMV Technologies, Piacenza, Italy) and seven fields per chamber were analyzed with CASA (HTM IVOS Version 12; IMV Technologies) using the standard setup for equine. The system parameter settings were: 45 frames acquired at 60 frames ⁄s; minimum contrast 70, minimum cell size 4 pixels; lower VAP cut-off 20 µm ⁄s, VAP cut-off for progressive cells 50 µm ⁄s and straightness 75%. Total motility, progressive motility, average path velocity and the number of rapid spermatozoa were recorded [22]. Those ejaculates containing >60% motile spermatozoa after dilution were used for subsequent assessment. Sixteen ejaculates were analyzed. Subsequently, each sample was fractionated in two aliquots: one was chilled at 4°C, the other one was frozen in 0.5 ml straw. The cooled-semen was placed in commercial styrofoam box and immediately shipped to the laboratory of Department of Emergency and Organs Transplantation (DETO) at the University of Bari, for immediate evaluation, while straws of frozen semen were stored in liquid nitrogen for successive evaluations.
Evaluation of plasma, acrosomal membrane and mitochondrial function
The fluorescent probes: H258 and CTC allow to evaluate the integrity of the plasmatic and mitochondrial membranes respectively. The first dye enters the cell and stains the nucleus when plasma membrane is damaged while the second enters the cell when the acrosomal membrane is damaged. Mitochondrial status was assessed by the lipophilic cationic JC-1 that differentiates mitochondria with low or high membrane potential by colors.
A stock solution of H258 was prepared to dissolve 10 mg in 100 µl distilled water. It was wrapped in foil and kept at 4°C. For use, 1 µl of the stock solution was diluted with 10 ml protein-free medium (PBS) and kept at +4°C. The fixative was made by mixing 1: 1 (v/v) 25% glutaraldehyde and 1 M Tris, pH 7.4 [23].
The CTC solution was made of 0.75 mM CTC and 5 mM L-cysteine in chilled CTC buffer containing 20 mM Tris and 130 mM NaCl, pH 7.8 [24].
The stock solution of 1.53 mM JC-1 in DMSO was prepared prior to use [16]. Different fluorescent probes with characteristics of excitation/emission were used simultaneously.
For each sample, aliquots of refrigerated and cryopreserved semen, pre-warmed at 37°C and to a final concentration of about 20×106 spermatozoa/ml, were suspended in 1 ml of Tyrode’s medium and JC-1 solution (2 µM final concentration) was added. Cells were incubated for 15 min at 37°C, then washed in PBS and incubated again for 2 min with 2 µl H258 solution. Unbound dye was removed centrifuging (900xg for 5 min) the stained suspension on 500 µl of 2% (w/v) polyvinylpyrrolidone in PBS.
The supernatant was discarded and the pellet re-suspended in 45 µl Tyrode’s medium. To each sample, 45 µl of the CTC solution and 4 ml of fixative solution were added and mixed. Few droplets of this suspension containing fixed and stained spermatozoa were placed between two coverslips and visualized by a Quanticell micro spectrofluorimetric system (VisiTech International, Sunderland, UK), consisting of a video digital imaging apparatus for the acquisition of color images (Nikon Instruments).
Plasma membrane integrity was evaluated using a 346-460 nm wavelength excitation/emission filters. Acrosomal membrane integrity was assessed by CTC at the 392-536 nm wavelength (excitation/emission). In mitochondria with high membrane potential, JC-1 forms multimeric aggregates and when excited at 480 nm, it emits light at 590 nm (high orange), on the contrary within mitochondria with low membrane potential, JC-1 forms monomers emitting at 525-530 nm (green) when excited at 488 nm wavelength.
For each ejaculate, about two hundred spermatozoa were analyzed and classified according to their florescence emission.
Sperm motility
Sperm kinetic was evaluated using a Sperm Analyzer (CASA-system; HTM-IVOS, Version 12.3, Hamilton-Thorne, Biosciences, MA, USA). Each ejaculate was analyzed for the cooled and frozen aliquots. Briefly, a 2 µl drop was recovered and placed in a Leja 4 analysis chamber, 20 micron depth (Leja Products B.V., The Netherlands). The analysis was performed at 37°C. The following motility characteristics were measured for each sample: percentage of motile spermatozoa; percentage of spermatozoa with a progressive motility; average path velocity (VAP): the velocity of the smoothed cell path (µm/s); straight line velocity (VSL): the average velocity measured in a straight line from the beginning to the end of the track (µm/s); curvilinear velocity (VCL): the average velocity measured over the actual point to point track followed by the cell (µm/s); amplitude of lateral head displacement (ALH); beat cross frequency (BCF); linearity (LIN); straightness (STR) and the percentage of rapid, medium, slow and static spermatozoa.
It has been necessary to set up the software as previously indicated, to clearly identify all spermatozoa, moreover to be sure that all sperm trajectories were correctly analyzed by CASA, the playback mode was also activated. Seven microscopic fields, selected randomly, were scanned.
Statistical analysis
The analysis of variance (ANOVA) was used to evaluate all recorded parameters. Differences were considered significant when P< 0.05. Linear regression and correlations among variables were also calculated.
Results
Evaluation of plasma, acrosomal membrane and mitochondrial function
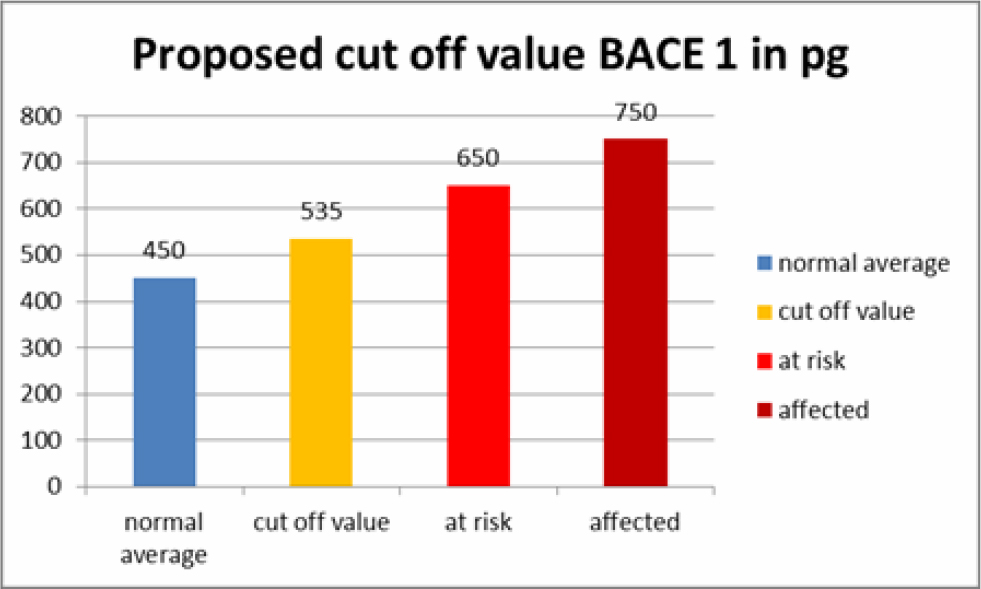
At first, in each sample (cooled and frozen), the integrity of both the plasmatic and acrosomal membrane were evaluated recording the percentage of male gametes with intact or damaged membrane, moreover the percentage of spermatozoa with high or low mitochondrial potential was also recorded (Figure 1).
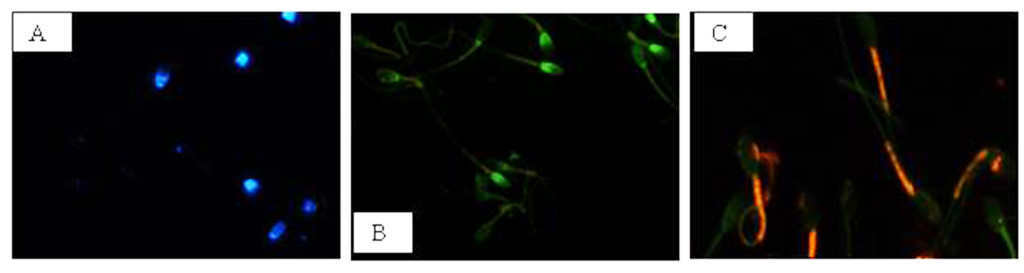
Figure 1. Photomicrography of equine spermatozoa. A Spermatozoa stained with H258 (Hoechst 33258), B Spermatozoa stained with CTC (chlortetracycline), C Spermatozoa stained with JC-1 (5,5’,6,6’-tetrachloro-1,1’,3,3’ tetraethylbenzimidazolyl carbocyanine iodide).
NIS-Elements F 3.0 (Nikon Instrument) software was employed to evaluate simultaneously all fluorescent probes. The results were used to classify spermatozoa in eight classes (Table 1), according to the morpho-functional state of plasmatic and acrosomal membranes and to mitochondrial function (Figure 2).
Table 1. Classification of equine sperm cells according to fluorescence emitted by H258, CTC and JC-1 probes.
|
Sperm Cell Category
|
aH258
|
bCTC
|
cJC-1
|
|
Intact plasma membrane, intact acrosome and high mitochondrial potential
Intact plasma membrane, intact acrosome and low mitochondrial potential
Intact plasma membrane, damage acrosome and high mitochondrial potential
Intact plasma membrane, damage acrosome and low mitochondrial potential
Damage plasma membrane, intact acrosome and high mitochondrial potential
Damage plasma membrane, intact acrosome and low mitochondrial potential
Damage plasma membrane, damage acrosome and high mitochondrial potential
Damage plasma membrane, damage acrosome and low mitochondrial potential
|
–
–
–
–
+
+
+
+
|
–
–
+
+
–
–
+
+
|
+
–
+
–
+
–
+
–
|
aH258 positive (+) = blue stained nucleus. bCTC positive (+) = green acrosome region. cJC-1 positive (+) = red in mid-piece region; negative (-) = green in mid-piece region.
Figure 2. Photomicrography of equine spermatozoa stained with H258,CTC and JC-1. A) Intact plasma membrane, intact acrosome and high mitochondrial potential, B) Intact plasma membrane, intact acrosome and low mitochondrial potential, C) Intact plasma membrane, damaged acrosome and high mitochondrial potential, D) Intact plasma membrane, damaged acrosome and low mitochondrial potential, E) Damaged plasma membrane, intact acrosome and high mitochondrial potential, F) Damaged plasma membrane, intact acrosome and low mitochondrial potential, G) Damaged plasma membrane, damaged acrosome and high mitochondrial potential, H) Damaged plasma membrane, damaged acrosome and
low mitochondrial potential.
The analysis revealed that both cooled and frozen-thawed samples had similar amount of spermatozoa with intact or damaged plasma membrane, also the percentage of cells with intact and damaged acrosomal membrane was not statistically different; on the contrary a dramatic decrease in the percentage of spermatozoa with high mitochondrial potential was observed in frozen samples (Table 2).
Table 2. Recovery rate: cooled versus frozen semen
|
Sperm parameters
|
cooled semen
|
Frozen semen
|
|
Intact plasma membrane (%)
Damage plasma membrane (%)
|
64
36
|
62
38
|
|
Intact acrosome (%)
Damage acrosome (%)
|
55
45
|
50
50
|
|
High mitochondrial potential (%)
Low mitochondrial potential (%)
|
47
53
|
24
76
|
Recovery rate (%) of spermatozoa showing different morphology of the acrosome and plasma membranes and mitochondrial potential in cooled and frozen semen.
Compared to cooled semen, in cryopreserved samples we recorded a reduction in the number of spermatozoa with intact plasma membrane, intact acrosome and high mitochondrial potential (6%); spermatozoa with intact plasma membrane, damaged acrosome and high mitochondrial potential were also lower (16%), as well as those with damaged plasma membrane, intact acrosome and high mitochondrial potential (1%). Moreover, also the number of spermatozoa with the damaged plasma membrane, intact acrosome and low mitochondrial potential were slightly reduced in frozen samples. While differences described above were not statistically significant, the reduction, caused by cryopreservation, in the class of intact plasma membrane, damaged acrosome and high mitochondrial potential spermatozoa was statistically significant (P< 0.001) as well as in the class of damaged plasma membrane, intact acrosome and low mitochondrial potential spermatozoa (P< 0.05) (Table 3).
Table 3. Cooled versus frozen semen.
|
Sperm Cell Category
|
Cooled semen
|
Frozen semen
|
|
Intact plasma membrane, intact acrosome and high mitochondrial potential
|
21%
|
15%
|
|
Intact plasma membrane, intact acrosome and low mitochondrial potential
|
9%
|
18%
|
|
Intact plasma membrane, damage acrosome and high mitochondrial potential
|
21%
|
5%
|
|
Intact plasma membrane, damage acrosome and low mitochondrial potential
|
13%
|
26%
|
|
Damage plasma membrane, intact acrosome and high mitochondrial potential
|
3%
|
2%
|
|
Damage plasma membrane, intact acrosome and low mitochondrial potential
|
22%
|
14%
|
|
Damage plasma membrane, damage acrosome and high mitochondrial potential
|
2%
|
2%
|
|
Damage plasma membrane, damage acrosome and low mitochondrial potential
|
9%
|
18%
|
Recovery rate of spermatozoa (%) for each of the eight morphological cell classes.
Cryopreservation led to an increase of spermatozoa with intact plasma membrane, intact acrosome and low mitochondrial potential spermatozoa (9%), of intact plasma membrane, damaged acrosome and low mitochondrial potential spermatozoa (13%), of damaged plasma membrane, damaged acrosome and low mitochondrial potential spermatozoa (9%), compared to refrigeration. These increases due to cryopreservation were statistically significant for intact plasma membrane, intact acrosome and low mitochondrial potential spermatozoa (P<0.05), for intact plasma membrane, damaged acrosome and low mitochondrial potential spermatozoa (P< 0.05) and for damaged plasma membrane, damaged acrosome and low mitochondrial potential spermatozoa (P< 0.05).
Sperm motility
Motility analysis was also conducted on each ejaculate of fresh semen; the recorded mean values of total and progressive motility were 79.06% ± 3.8 and 63.87% ± 2.16, respectively.
Sperm motility values of cooled and frozen-thawed semen are shown in Table 4. Semen cryopreservation affected sperm motility. Refrigerated semen displayed higher total and progressive motility than frozen-thawed semen (P< 0.001). The estimated reduction in the percentage of total and progressive motile spermatozoa related to cryopreservation was 20.33% and 19.57%, respectively.
Table 4. Motility parameters: cooled versus frozen semen.
|
Motility parameters
|
cooled semen
|
frozen semen
|
|
Total motility (%)
Progressive motility (%)
VAP (mm/s)
VSL (mm/s)
VCL (mm/s)
ALH (mm)
BCF (Hz)
STR (%)
LIN (%)
Rapid spermatozoa (%)
Medium spermatozoa (%)
Slow spermatozoa (%)
Static spermatozoa (%)
|
74,42 ± 3,69 **
59,37 ± 1,76 **
108,71 ± 17,10 **
58,31 ± 13,04
224,44 ± 30,70 **
8,24 ± 0,94 **
34,61 ± 5,18
53,75 ± 10,66
26,86 ± 6,26
45,61 ± 11,05 **
7,36 ± 3,43
7,16 ± 3,32
39,88 ± 11,69
|
54,09 ± 3,43
39,8 ± 3,09
73,48 ± 15,37
57,8 ± 12,87
149,83 ± 21,24
6,21 ± 0,65
39,28 ± 3,24
71,91 ± 5,71
38,96 ± 4,41
16,79 ± 10,78
8,41 ± 5,95
13,99 ± 7,47 *
61,22 ± 20,84 *
|
Motility parameters of equine cooled or frozen sperm samples: VAP (average path velocity); VSL (straight line velocity); VCL (curvilinear velocity); ALH (amplitude of lateral head displacement); BCF (beat cross frequency); STR (straightness); LIN (linearity). * P < 0.05, ** P < 0.001.
Evaluated VAP and VCL were lower in frozen-thawed semen than in refrigerated one (P< 0.001).
The VSL of refrigerated spermatozoa did not differ significantly from that of cryopreserved sperm.
The ALH was higher in cooled than in frozen-thawed semen (P<0.001). The BCF of cooled spermatozoa did not differ significantly from that of frozen sperm cells.
Cooled semen displayed a higher percentage of rapid spermatozoa (P<0.001) while a 28.82% reduction was induced by cryopreservation. The percentage of medium spermatozoa was similar in cooled and frozen semen. Slow and static spermatozoa were higher in frozen-thawed than in cooled semen (P<0.05) the percentage increase, probably due to the cryopreservation process, was 6.83% and 21.34%, respectively.
Correlations
Significant correlations were observed between mitochondrial energy level and different sperm membrane conditions. In cooled semen a high correlation between high mitochondrial potential and intact plasma membrane (r=0.91), high mitochondrial potential and intact acrosome (r=0.93), low mitochondrial potential and damage acrosome (r=0.90), were found. In frozen semen, a high correlation between low mitochondrial potential and damaged plasma membrane (r=0.87), low mitochondrial potential and damaged acrosome (r=0.92) were found. On the contrary, no correlations between mitochondrial membrane potential and different motility characteristics were evidenced.
Discussion
Severe changes in temperature are a common feature of semen preservation protocols but are not a biological phenomenon to which the sperm cell is adapted [25]. Temperature transitions associated with semen fast chilling or freezing are in fact well known for their production of deleterious effects on sperm survival and consequently, lower conception rates following artificial insemination [5].
The reduction in fertilizing ability has typically been attributed to a reduced rate of sperm motility and to the induction of morphological abnormalities [5]. Damages to the plasma membrane, to the acrosomal membrane and to the mitochondrial function of spermatozoa undergoing cooling and/or freezing depend on changes of temperatures and osmolarity, which cause morphological alterations in the organization and composition of proteins and lipids of the sperm surface [26].
Sperm quality related to functional characteristics has been investigated by the association of different fluorescent probes in ram [27], bull [28] and stallion [13, 16, 18].
In this study we validated the association of three fluorescent probes H258, CTC and JC-1 to evaluate the state of plasma and acrosomal membranes, as well as the mitochondrial function both in cooled and frozen equine sperm, using the inverted fluorescent microscopy highlighting the prominent role of the mitochondrion as marker for equine sperm quality.
We found both in cooled and frozen equine semen samples a higher number of spermatozoa with intact plasma membranes compared to those with destroyed membranes, this result is in line with the observations reported by Vasconcelos et al., [29] and by De Leeuw et al., [30].
It is reasonable to attribute the poor fertility of frozen semen to ultrastructural modifications of the cell membranes and to phase transitions occurring during cooling and rewarming. It seems reasonable that the reorganization of sperm membrane bilayer alters the interactions among lipids and between lipids and proteins required for normal membrane functions, determining a re-modelling of the membrane components [31, 32] and the decreased amount of polyunsaturated fatty acids and cholesterol [33]; [34]. Plasma membrane disruption due to cooling or freezing determines the loss of cations and enzymes from the sperm cells, providing some explanation for the reduction in sperm motility and metabolic activity observed in cold-shock spermatozoa [35]. Cold shock also destroys the selective permeability of sperm membranes to calcium, thus leading to its excessive intracellular accumulation, and consequently, to a reduction in motility and cell necrosis. These processes result in an advanced stage of sperm maturation characterized by capacitation, hyperactivation and acrosome reaction [35].
For what concerns the acrosome membrane, in the present study cooled spermatozoa displaying their integrity, were more numerous than those with a damaged acrosome membrane. As to mitochondrial membrane potential, it is a sensitive indicator of the functional status of the organelle; in the present study, cooled spermatozoa with intact plasma membrane showed a high mitochondrial potential. Whereas a high percentage of spermatozoa with an intact plasma membrane and low mitochondrial potential was observed among cryopreserved ones, suggesting that cryopreservation may result in a significant loss in mitochondrial potential.
Cryopreservation amplified the mitochondrial damage compared to cooling, probably because of the block in ATP synthesis and consequently in the reduction in the activity of ATP-dependent pumps [36], a tendency to make the plasma membrane unstable. This may lead to an increase in intracellular Na+ and a reduction in intracellular K+, with reduced Ca2+ efflux due to inhibition of the Ca2+ ATP-ase pumps, increased leakage of Ca2+ across the membrane due to the instability caused by removal of cholesterol, and/or increased Ca2+ influx due to the activation of unidentified channels [37]. Changes associated with this process include an increase of capacitated and acrosome-reacted spermatozoa, as reported by Felix [37]. These results are in agreement with those reported by Albrizio et al., [38]; they showed that cryopreservation increases the percentage of capacitated and acrosome reacted spermatozoa, increase the intracellular Ca2+ concentration and leads to a higher functional response of L-type Ca2+-channels to both the agonist (Bay K-8644) and antagonist (Nifedipine). Moreover, cryopreservation was found to determine the activation of lytic enzymes and to increase mitochondrial permeability, leading to cell death [10]. This is in accordance with the results of the present study, in which the percentage of spermatozoa with damaged acrosome membrane and low mitochondrial potential is higher in frozen than in cooled ones.
It has been suggested that plasma membranes and mitochondria in bovine sperm are both affected by cryopreservation [39]. In accordance with the above-mentioned study, we found that plasma membrane and mitochondrial membrane were equally vulnerable to the freezing and thawing process, because of their high thermolability [40]. Moreover, in this study, plasma membrane and mitochondrial membrane conditions are highly correlated demonstrating that these structures depend on each other, as reported by Celeghini et al., [7]. The mitochondrial plasma membrane is a good predictor to determine and assess plasma membrane and acrosome membrane damages because of thermic treatment. Besides structural sperm characteristics, conflicting opinions concerning the relationship between motility and fertility in the stallion exist [41, 42] although sperm cell motility and the quality of motility are still the most reliable indicators of sperm viability. In this study, total motility recorded in cooled semen was in agreement with similar data reported by Love et al.,[13] and Heckenbichler et al., [43], whereas, the same parameter found in frozen semen was similar to that found by Wrench et al., [44] and Salazar et al., [45]. The progressive motility found in cooled semen was similar to that reported by Cocchia et al., [46] and Heckenbicher et al., [43], whereas the same parameter found in frozen semen was similar to that found by Wrench et al., [44]. It is well known the functional relationship between the mitochondrion and the cellular apparatus that drives sperm movements; in fact mitochondria are well known for producing energy in the form of ATP and sperm motility depends upon the energy produced by the oxidative phosphorylation. Mitochondria are a major source of Reactive Oxygen Species (ROS) and appear to be the cellular structures most susceptible to damages during cooling and cryopreservation [47], leading to a loss of motility in frozen semen.
As to VCL, VAP, VSL and ALH parameters, the results found in cooled semen were in agreement with those of Aurich et al., [48], whereas those found in frozen semen were similar to the results of Salazar et al., [45].
This study demonstrates that the quality of motility is different in cooled and frozen semen, in fact, cooled samples showed higher VAP and VCL values than frozen ones. These results suggest that motile cells, that are numerically higher in cooled than in frozen semen, are also characterized by higher speed as deduced by their higher VAP and VCL, on the contrary, linear characteristic of sperm motility is not affected by temperature reduction. We found that ALH values were higher in refrigerated semen than in the cryopreserved one. In general, higher ALH values are unwanted, because they can interfere with cell progression, as demonstrated by [18], which stand for reduced sperm quality. However until now a cutoff value for ALH has not been defined so that is not clear the influence of this parameter on the progressive motility and on the ability of the spermatozoon to go forward into the female reproductive tract [7].
As to motility characteristics, other authors found opposite results for refrigerated and cryopreserved semen [42]; [49]; [50]. This could be only an apparent discordance considering that many factors may have a role such as season of semen recovery, physiological differences among stallions, frequency in collecting semen, different extenders for semen dilutions, different settings of the CASA system (threshold setting, specimen concentration, video digitalization rate) [9, 17, 51-53].
We, as several authors working on other animal species, didn’t find a direct correlation between mitochondrial function and a specific kinetic parameter, probably because equine sperm motility is modulated by different factors not only by mitochondria [14, 23].
In conclusion, our study, reporting the simultaneous evaluation of different functional characteristics of equine spermatozoa subjected to temperature changes, increases the information on semen quality helping the selection of those characteristics more correlated with semen fertility potential. In particular, we demonstrated that mitochondria are good indicators of sperm survival and can be used to discriminate between good and poor semen quality. Moreover, they are strictly correlated to the state of cellular membrane, therefore, a proper evaluation of mitochondria sounds necessary when analyzing sperm samples. Standing on the present study, mitochondrial membrane potential provides useful information on equine sperm quality so that it may be exploitable as a marker of sperm fertility; moreover, the high mitochondrial sensitivity to temperature variations observed in this study highlights the importance of developing new strategies to protect the functionality of this organelle to preserve sperm cell physiology.
Disclosure
None of the authors of this paper has a financial or personal relationship with other people or organizations that could inappropriately influence or bias the content of the paper.
This paper was concepted by GM Lacalandra, G. Mari and A. Zarrilli; written by M. Albrizio, G. Mari, GM Lacalandra and A. Zarrilli. Data were acquired by AC Guaricci, A. Moramarco and E. Micera; B. Mislei, and G. Rizzato analysed and interpreted the data. Albrizio revised the manuscript and cared the journal submission.
All authors approved the final article.
Acknowledgements: This work was supported by Italian University funds
References
- Allen WR (2005) The development and application of the modern reproductive technologies to horse breeding. Reprod Domest Anim 40: 310–29.
- Macías García B, González Fernández L, Ortega Ferrusola C, Morillo Rodríguez A, Gallardo Bolaños JM, et al. (2011) acids and plasmalogens of the phospholipids of the sperm membranes and their relation with the post-thaw quality of stallion spermatozoa. Theriogenology 75: 811–8.
- Aurich C (2005) Factors affecting the plasma membrane function of cooled-stored stallion spermatozoa. Anim Reprod Sci 89: 65–75.
- Leahy T(2011) Gadella BM. Sperm surface changes and physiological consequences induced by sperm handling and storage. Reproduction 142: 759–78.
- Holt WV (2000) Basic aspects of frozen storage of semen. Anim Reprod Sci 62: 3–22. [crossref]
- Anzar M, He L, Buhr MM, Kroetsch G, Pauls KP (2002) Sperm apoptosis in fresh and cryopreserved bull semen detected by flow cytometry and its relationship with fertility. Biol Reprod 66: 354–60.
- Celeghini ECC, de Andrade AFC, Raphael CF, Nascimento J, Ticianelli JS, et al. (2010) Damage assessment of the equine sperm membranes by fluorimetric tecnique. Braz Arch Biol Techn 53: 1285–92.
- Colenbrander B, Gadella BM, Stout TAE (2003) The predictive value of semen analysis in the evaluation of stallion fertility. Reprod Dom Anim 38: 305–11.
- Giannoccaro A, Lacalandra GM, Filannino A, Pizzi F, Nicassio M, et al. (2010) Assessment of viability, chromatin structure stability, mitochondrial function and motility of stallion fresh sperm by using objective methodologies. J Cell Anim Biol 4: 34–41.
- Neild DM, Gadella BM, Chaves MG, Miragaya MH, Colenbrander B, Aguero A (2003) Membrane changes during different stages of a freeze-thaw protocol for equine semen cryopreservation. Theriogenology 59: 1693–705.
- Rodriguez-Martines H (2003) Laboratory semen assessment and prediction of fertility: still utopia? Reprod Dom Anim 38: 312–8.
- Gillan L, Evans G, Maxwell WMC (2005) Flow cytometric evaluation of sperm parameters in relation to fertility potential period. Theriogenology 63: 445–57.
- Love CC, Thompson JA, Brinsko SP, Rigby SL, Blanchard TL, Lowry VK Varner DD (2003) Relationship between stallion sperm motility and viability as detected by two fluorescence staining techniques using flow cytometry. Theriogenology 60: 1127–38.
- Martinez-Pastor F, Johannisson A, Gil J, Kaabi M, Anel L, Paz P, Rodriguez-Martinez H (2004) Use of chromatin stability assay, mitochondrial stain JC-1, and fluorometric assessment of plasma membrane to evaluate a frozen-thawed ram semen. Anim Reprod Sci 84121–33.
- Flesch FM, Gadella BM (2000) Dynamics of the mammalian sperm plasma membrane in the process of fertilization. Biochim Biophys Acta 1469: 197–235.
- Gravance CG, Garner DL, Baumber J, Ball BA (2000) Assessment of equine sperm mitochondrial function using JC-1. Theriogenology 53: 1691–703.
- Ball BA, Medina V, Gravance CG, Baumber J (2001) Effect of antioxidants on preservation of motility, viability and acrosomal integrity of equine spermatozoa during storage at 5 °C. Theriogenology 56: 577–89.
- Arruda RP, Ball BA, Gravance CG, Liu IKM (2003) Flow cytometric membrane and acrosomal integrity of the stallion spermatozoa. Acta Sci Vet 31: 226–7.
- Tartaglione CM, Ritta MN (2004) Prognostic value of spermatological parameters as predictors of in vitro fertility of frozen-thawed bull semen. Theriogenology 62: 1245–52.
- Rathi R, Colenbrander B, Bevers MM, Gadella BM (2001) Evaluation of in vitro capacitation of stallion spermatozoa. Biol Reprod 65: 462–70.
- Kenney RM, Bergman RV, Cooper WL, Morse GW (1975) Minimal contamination techniques for breeding mares: techniques and preliminary findings. Proc Am Ass Equine Pract 21: 327–36.
- Mari G, Rizzato G, Merlo B, Iacono E, Seren E, Tamanini C, et al. (2008) uality and fertilizing ability in vivo of sex-sorted stallion spermatozoa. Reprod Dom Anim 45: 331–5.
- Albrizio M, Guaricci AC, Maritato F, Sciorsci RL, Mari G, et al. (2005) Expression and subcellular localization of the mu-opioid receptor in equine spermatozoa: evidence for its functional role. Reproduction 129: 39–49.
- Neild DM, Gadella BM, Aguero A, Stout TAE, Colenbrander B (2005) Capacitation, acrosome function and chromatin structure in stallion sperm. Anim Reprod Sci 89: 47–56.
- Lopez-Fernandez C, Crespo F, Arroyo F, Fernandez JL, Arana P, et al. (2007) Dynamics of sperm DNA fragmentation in domestic animals II. The stallion. Theriogenology 68: 1240–50.
- Amann RP, Pickett BW (1987) Principles of cryopreservation and a review of cryopreservation of stallion spermatozoa. J Equine Vet Sci 7: 145–73.
- Windsor DP (1997) Mitochondrial function and ram sperm fertility. Reprod Fertil Dev 9: 279–84.
- Thomas CA, Garner DL, Dejarnette JM, Marshall CE (1997) Fluorometric assessments of acrosomal integrity and viability in cryopreserved bovine spermatozoa. Biol Reprod 56: 991–8.
- Vasconcelos AB, Santana MA, Santos AMC, Santoro MM, Lagares MA (2011) Metabolic evaluation of cooled equine spermatozoa. Andrologia 42: 106–11.
- Heise A, Thompson PN, Gerber D (2011) Influence of seminal plasma on fresh and post-thaw parameters of stallion epididymal spermatozoa. Anim Reprod Sci 123: 192–201. [crossref]
- De Leeuw FE, Chen H-C, Colenbrander B, Verkleij AJ (1990) Cold-induced ultrastructural changes in bull and boar sperm plasma membranes. Cryobiology 27: 171–83.
- Drobnis EZ, Crowe LM, Berger T, Anchordoguy TJ, Overstreet JW, Crowe JH (1993) Cold shock damage is due to lipid phase transitions in cell membranes: a demonstration using sperm as a model. J Exp Zool 265: 432–7.
- Maldjian A, Pizzi F, Gliozzi T, Cerolini S, Penny P, et al. (2005) Changes in sperm quality and lipid composition during cryopreservation of boar semen. Theriogenology 63: 411–421. [crossref]
- Chakrabarty J, Banerjee D, Pal D, De J, Ghosh A Majumder GC (2007) Shedding off specific lipid constituents from sperm cell membrane during cryopreservation. Cryobiology 54: 27–35.
- Bailey JL, Bilodeau JF, Cormier N (2000) Semen cryopreservation in domestic animals: a damaging and capacitating phenomenon. J Androl 21: 1–7. [crossref]
- Silva PF, Gadella BM (2006) Detection of damage in mammalian sperm cells. Theriogenology 65: 958–978. [crossref]
- Felix R (2005) Molecular physiology and pathology of Ca2+-conducting channels in the plasma membrane of mammalian sperm. Reproduction 129: 251–62.
- Albrizio M, Lacalandra GM, Micera E, Moramarco AM, Surdo NC, et al. (2010) Effects of cryopreservation on capacitation, acrosome reaction and activity of L-type voltage-dependent calcium channels of equine sperm cells. Proceedings of the Tenth International Symposium on Equine Reproduction, Lexington; KY, USA. Anim Reprod Sci 121: 218–19.
- Bollwein H, Fuchs I, Koess C (2008) Interrelationship between plasma membrane integrity, mitochondrial membrane potential and DNA fragmentation in cryopreserved bovine spermatozoa. Reprod Dom Anim 43: 189–95.
- Breininger E, Beorlegui NB, O’Flaherty CM, Beconi MT (2005) Alpha-tocopherol improves biochemical and dynamic parameters in cryopreserved boar semen. Theriogenology 63: 2126–35.
- Jasko DJ, Hataway JA, Schaltenbrand VL, Simper WB, Squires EL (1992) Effect of seminal plasma and egg yolk on motion characteristics of cooled stallion spermatozoa. Theriogenology 37: 1241–52.
- Kuisma P, Andersson M, Koskinen E, Katila T (2006) Fertility of frozen-thawed stallion semen cannot be predicted by the currently used laboratory methods. Acta Vet Scand 48: 14–22.
- Heckenbichler S, Deichsel K, Peters P, Aurich C (2011) Quality and fertility of cooled-shipped stallion semen at the time of insemination. Theriogenology 75: 849–56.
- Wrench N, Pinto CR, Klinefelter GR, Dix DJ, Flowers WL, et al. (2010) Effect of season on fresh and cryopreserved stallion semen. Anim Reprod Sci 119: 219–227. [crossref]
- Salazar JL Jr, Teague SR, Love CC, Brinsko SP, Blanchard TL, et al. (2011) Effect of cryopreservation protocol on postthaw characteristics of stallion sperm. Theriogenology 76: 409–418. [crossref]
- Cocchia N, Pasolini MP, Mancini R, Petrazzuolo O, Cristofaro I, et al. (2011) Effect of sod (superoxide dismutase) protein supplementation in semen extenders on motility, viability, acrosome status and ERK (extracellular signal-regulated kinase) protein phosphorylation of chilled stallion spermatozoa. Theriogenology 75: 1201–10.
- Ortega-Ferrusola C, Garcia BM, Gallardo-Bolanos JM, Gonzalez-Fernandez L, Rodriguez-Martinez H, et al. (2009) Apoptotic markers can be used to forecast the freezeability of stallion spermatozoa. Anim Reprod Sci 114: 393–403.
- Aurich C, Seeber P, Muller-Schlosser F (2007) Comparison of Different Extenders with Defined Protein Composition for Storage of Stallion Spermatozoa at 5°C. Reprod Dom Anim 42: 445–8.
- Rota A, Furzi C, Panzani D, Camillo F. Studies on Motility and Fertility of Cooled Stallion Spermatozoa. Reprod Dom Anim 39: 103–9.
- Spizziri BE, Fox MH, Bruemmer JE, Squires EL, Graham,JK (2010) Cholesterol-loaded-cyclodextrins and fertility potential of stallions spermatozoa. Anim Reprod Sci 118: 255–64.
- Brinsko SP, Crockett EC, Squires EL (2000) Effect of centrifugation and partial removal of seminal plasma on equine spermatozoal motility after cooling and storage. Theriogenology 54: 129–36.
- Sieme H, Katila T, Klug E (2004) Effect of semen collection practices on sperm characteristics before and after storage and on fertility of stallions. Theriogenology 61: 769–84.
- Pena FJ, Macías García B, Samper JC, Aparicio IM, Tapia JA, et al. (2011) Dissecting the molecular damage to stallion spermatozoa: The way to improve current cryopreservation protocols? Theriogenology 76: 1177–86.